A Method for Effectively Overcoming Tight Functional Linkage Between Genes in Rice by CRISPR/Cas9 System
Anthocyanins are widely distributed in one or more parts of rice (Oryza sativa L.) plants, including seed coat, stigma, apiculus, leaf sheath and leaf blade, and are the main pigments used in rice to achieve different colors (Hou et al, 2009; Aizza and Dornelas, 2011). In rice, tissue-specific color traits (especially the color of apiculus, namely the lemma and palea of the spikelet) are not only important for rice variety identification but also important for linkage analysis and rice domestication research (Saitoh et al, 2004; Fan et al, 2007; Lin et al, 2019). The apiculus color is controlled by the complementary functions of three pairs of dominant genes, C, A and P. Gene C (chromogen) is a pigment gene, which is the basic gene for producing pigments. Gene A (activator) activates gene C, converting the chromogen into anthocyanins, and gene P (purple) controls the distribution of anthocyanins in various organs (Reddy, 1996; Sakamoto et al, 2001). OsC1, which determines apiculus color, locates on chromosome 6 (Zhao et al, 2016). S5n, which controls wide compatibility in crosses between rice subspecies, locates 427 kb ± 94 kb away from OsC1 in physical distance on chromosome 6, and is genetically tightly linked with OsC1 (Sheng et al, 2008). In general, there is genetic linkage between desirable and undesirable genes, in which case, if the desirable gene is introduced into recurrent parents by backcrossing, the undesirable gene would be also introduced. This genetic phenomenon is regarded as linkage drag. In breeding, it is difficult to break linkage drag by selecting for rare recombinants, especially for tightly linked genes. Therefore, a new method is needed to address this problem. CRISPR/Cas9 system provides a convenient and powerful tool for targeted genome editing in different species, and it has been widely used in rice for improvement of a range of traits (Zhou et al, 2016; Shao et al, 2017; Sun et al, 2017; Tang et al, 2017; Zhang et al, 2018). In Arabidopsis, a double mutant of closely linked genes M320 and M330 has been created using the CRISPR/Cas9 system (Pang et al, 2018). In the current study, our goal was to overcome the tight linkage between OsC1 and S5n using the CRISPR/Cas9 system to create new sterile line resources with wide-compatibility and colorless apiculus.
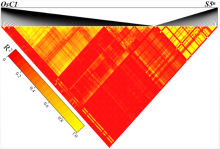
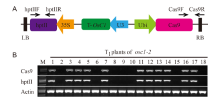
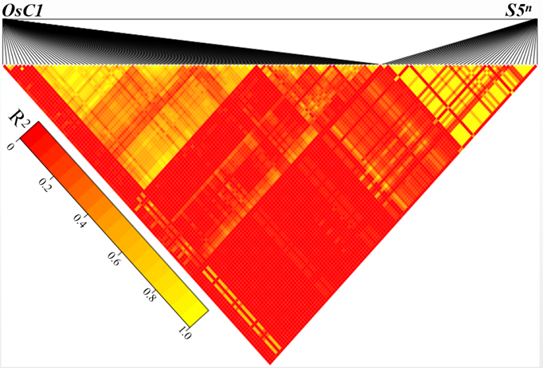
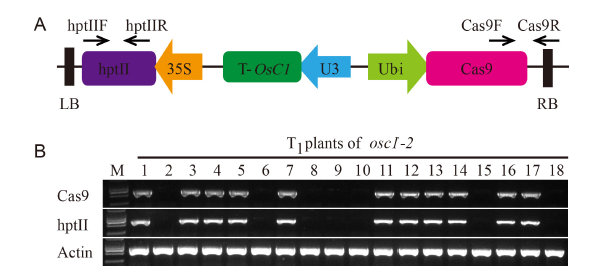
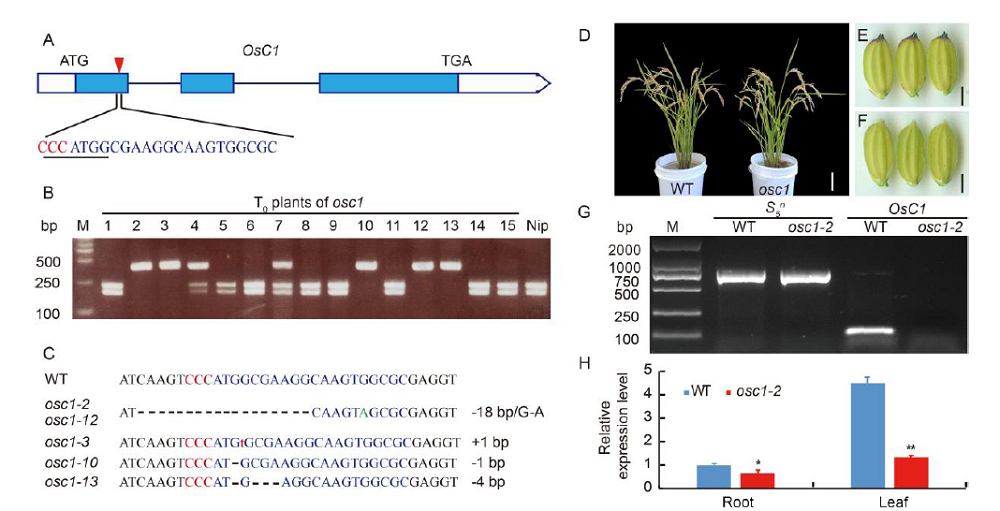
In this study, we confirmed that there was linkage disequilibrium between OsC1 and S5n within 132 japonica accessions (Supplemental Fig. 1). To overcome the tight functional linkage between the two genes, we constructed a pC1300-Ubi::Cas9-sgRNAOsC1 expression vector, with the target site being specifically chosen on exon 1 of OsC1in 4008S (a japonica-type wide-compatibility photosensitive genic male-sterile line with purple apiculi) (Fig. 1-A). The target contained restriction sites that were used for screening mutations using a PCR-based restriction enzyme (PCR/RE) digestion assay. T0 plants were screened for the presence of frame-shift mutations in OsC1, and five homozygous mutant plants (osc1-2, -3, -10, -12 and -13) were identified from 15 transgenic plants using the PCR/RE assay (Fig. 1-B). A total of four different mutation types were identified from the five homozygous mutant plants by the Sanger sequencing (Fig. 1-C). These four different mutation types all create a stop codon in the coding area, thus leading to the production of an immature protein of OsC1. In this study, we randomly chose the osc1-2 mutant to produce the T1 generation. Previous studies indicate that there must be a difference of at least two bases between the sgRNA and the target sequence if off-targeting events are to be avoided (Li et al, 2016). To determine whether off-targeting had taken place in the osc1-2 plant line, four putative off-target sites which showed high homology with the sgRNA of OsC1 were searched, and we found that none of the four putative off-target sites produced off-target events (Supplemental Table 1). To determine whether Cas9 and the hygromycin resistance gene (hptII) sequences were present in these mutant lines, PCR amplification was performed using the primers designed to specifically amplify Cas9 and hptII sequences, respectively (Supplemental Fig. 2-A). Seven possible transgene-free homozygous osc1-2mutant plants were isolated from 18 T1 plants (Supplemental Fig. 2-B). These possible transgene-free mutant T1 plants represented a new germplasm resource for rice breeding and were chosen for further studies.
Supplemental Table 1.
Supplemental Table 1.
 Supplemental Table 1. Mutations detected in putative CRISPR/Cas9 off-target sites.| Plant line was sequenced | Target | Gene | Off-target site | No. of mismatching bases | No. of plants sequenced | No. of plants with mutations | Mutation rate (%) |
|---|
| osc1-2 | OsC1 | Os06g0205100 | GAGCCAATGGCCTTCGCCATGGG | 3 | 36 | 0 | 0 | | | | CTCCCTCTTGCCTTCGCCATGGG | 4 | 36 | 0 | 0 | | | | GCGCCGCTGGCGTTCGCCATCGG | 3 | 36 | 0 | 0 | | | | CCGCCCATGGCCTTCGCCATAGG | 4 | 36 | 0 | 0 |
The PAM motif (NGG) is shown in green; mismatch bases are shown in red. | Supplemental Table 1. Mutations detected in putative CRISPR/Cas9 off-target sites. |
To determine the phenotypic pattern in the mutants, we examined the color trait of the wild type and osc1-2 in the apiculi. The wild type 4008S exhibited purple apiculi at the heading stage. However, osc1-2displayed colorless apiculi (Fig. 1-D to -F). To examine whether the tight functional linkage between OsC1 and S5n present in the wild type was overcome in the osc1-2 mutant, primers (S5n-JD-F/R and OsC1-JD-F/R) were designed to specifically amplify S5n and OsC1 sequences in the wild type and the osc1-2 mutant, respectively (Supplemental Table 2). The results indicated that the sequence ofS5n can be amplified in both the wild type and the osc1-2 mutant, whereas the sequence ofOsC1 can be amplified in only the wild type but not in osc1-2 (Fig. 1-G). In addition, we also measured the expression of OsC1 in different organs of wild type and osc1-2 by quantitative real-time PCR (qRT-PCR). The data indicated that OsC1was constitutively expressed in the root and particularly in the leaf of the wild type, whereas expression was significantly down-regulated in both organs of the mutant line (Fig. 1-H), which is consistent with the expression profile reported by Zhao et al (2016). These results indicated that, using the CRISPR/Cas9 system, we were able to overcome the co-inheritance of the wide-compatibility allele S5n and the purple apiculus allele OsC1in 4008S.
Supplemental Table 2.
Supplemental Table 2.
Supplemental Table 2. Primers used in this research.| Primer name | Sequence (5′ -3′ ) | Purpose |
|---|
OsC1-g++
OsC1-g--
T3
OsC1-JC-F
OsC1-JC-R
hptII F | GGCAGCGCCACTTGCCTTCGCCAT
AAACATGGCGAAGGCAAGTGGCGC
ATTAACCCTCACTAAAGGGA
CCAGATCGCTCAGTCTCACA
TAGGCCGGAGATAGTTGAGC
GCTGTTATGCGGCCATTGTC | Vector construction
Vector construction
Vector construction
Detection of target mutations
Detection of target mutations
Genotyping | | hptII R | GACGTCTGTCGAGAAGTTTC | Genotyping | | Cas9 F | ACCAGACACGAGACGACTAA | Genotyping | | Cas9 R | ATCGGTGCGGGCCTCTTC | Genotyping | | Actin-F | TGCTGACAGGATGAGCAAGG | Genotyping | | Actin-R | CCCAACCATGCAAAGCTCAC | Genotyping | | OsC1-qPCR-F | ATGGGGAGGAGAGCTTGCTG | qPCR | | OsC1-qPCR-R | CGGAGATAGTTGAGCCACC | qPCR | | Ubi-qPCR-F | GCTCCGTGGCGGTATCAT | qPCR | | Ubi-qPCR-R | CGGCAGTTGACAGCCCTAG | qPCR | | S5n-JD-F | CTTCATTCCCAGCGAGCGG | Genotyping | | S5n-JD-R | ATGGGCGGAGGCATTGGT | Genotyping | | OsC1-JD-F | GCAAAGGAAGGGATGAAGAG | Genotyping | | OsC1-JD-R | CGTCATCGCCGTCTCCTAATT | Genotyping |
| Supplemental Table 2. Primers used in this research. |
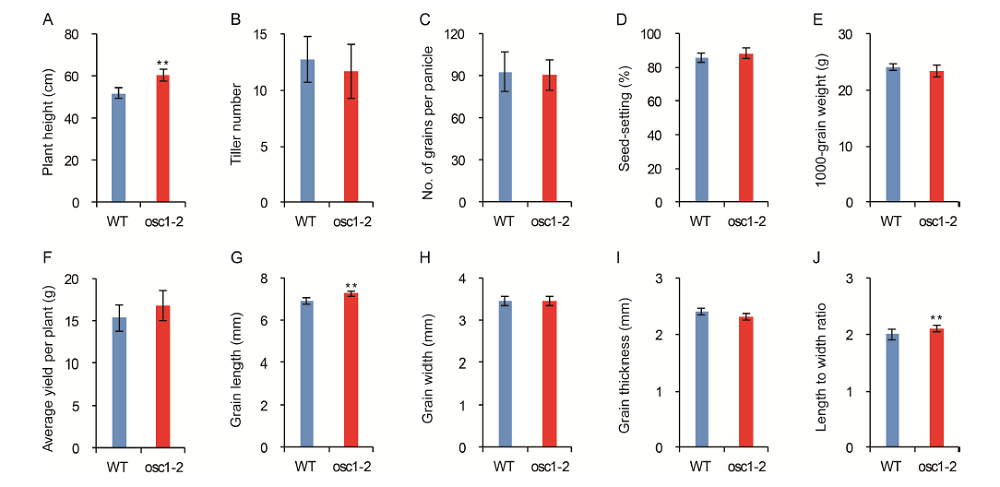
In addition to the apiculus color trait, we also investigated agronomic traits to determine whether there were any pleiotropic effects of the frame-shift mutation. The data indicated that the plants of the osc1-2 mutant were significantly higher than the wild type, but there was no significant difference in either the tiller number or the number of grains per panicle between the wild type and osc1-2 (Supplemental Fig. 3-A to -C). In addition, the seed-setting rate, the 1000-grain weight, and the average yield per plant of osc1-2 also exhibited no significant difference from those of the wild type (Supplemental Fig. 3-D to -F). The grain length of the mutant was significantly greater than that of the wild type, but there were no significant differences in the grain width and the grain thickness of the wild type and the mutant, meaning that the length-to-width ratio of the mutant was significantly higher than that of the wild type (Supplemental Fig. 3-G to -J). These results indicate that, except for apiculus color and plant height, the osc1-2 line exhibited normal agronomic traits relatively unchanged from the wild type. Overall, a frame-shift mutation of OsC1 in the genetic background of 4008S overcame the detrimental effect of the tight linkage between OsC1 and S5n, providing a new germplasm resource for rice breeders.
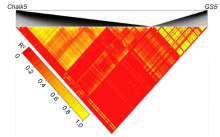
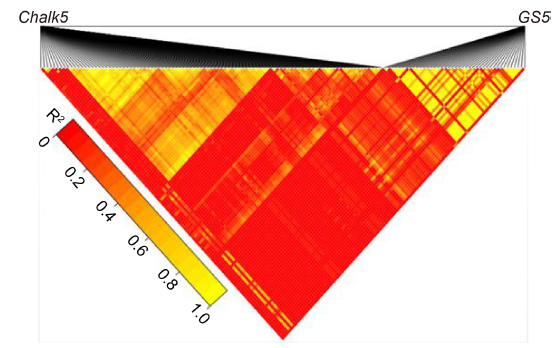
The purple apiculus trait controlled by OsC1 is an easy visual marker, and often used for varietal selection and evaluation of the purity of a seed population, which is tightly linked in coupling to the wide-compatibility allele S5n. Such tight gene linkage is common in rice and can be used for varietal improvement in rice breeding, as tight linkage of desirable alleles can be beneficial in the selection of improved varieties. For example, the two QTLs (qSPP5 for spikelet number per panicle and qTGW5 for grain weight) are tightly linked on chromosome 5, which can be valuable for improving rice yields (Luo et al, 2013). However, the tight functional linkage between desirable and undesirable genes is complicated and challenging in terms of its application in rice breeding. For example, the tight linkage between the positive QTLs GS5 and Chalk5 (Supplemental Fig. 4), which regulate grain size and white belly grain rate, respectively, is not conducive to the development of high-yielding and good-quality rice varieties (Li et al, 2011). Another example is the tight linkage between the desirable rice blast resistance gene and the undesirable high chalky gene, which causes a barrier in breeding of good-quality and high disease-resistant rice varieties (Fukuoka et al, 2009).
Aggregation of large numbers of desirable genes by hybridization is the basis for developing improved varieties by traditional breeding. However, this method has certain disadvantages. Firstly, depending on the crop and its breeding system, it takes more than a decade to hybridize and accumulate multiple desirable genes, and the workload is large. Secondly, when desirable and undesirable genes are tightly linked, it can be difficult to eliminate the undesirable gene by selection, which hinders the use of the desirable genes. Therefore, it is necessary to develop effective methods to overcome the tight linkage between desirable and undesirable genes.
The CRISPR/Cas9 system can precisely edit the sequence of any gene, with the advantages of simple operation, a low technical threshold and a short experimental period (Baltes and Voytas, 2015). In our study, the problem of the tight functional linkage between OsC1 and S5n was successfully overcome by mutating the purple apiculi allele into the colorless apiculus allele (Supplemental Fig. 1), resulting in the production of a valuable new genetic resource for breeding, the osc1mutant combined with the wide-compatibility gene S5n but with colorless apiculi. Furthermore, through self-pollination, the T-DNA sequences can be removed and possible transgene-free plants were generated from the T1 generation in only one year (Supplemental Fig. 2-B). Therefore, the CRISPR/Cas9 system is an effective method for rapidly breaking the linkage drags. Although genetic modification (GM) crops still have great controversy in China, genome editing is, in many ways, even more precise and predictable than transgenesis. Crops bred by genome editing are defined as genome-edited crops (GECs) and the US Department of Agriculture does not consider GECs to be GM organisms as long as GECs do not contain DNA from plant pests. Moreover, in many cases, GECs are more promising than those from conventional breeding, which would be offered more opportunities for ensuring global food and nutrition security (Huang et al, 2016). In a word, this strategy would have wider applications in significantly accelerating the use in breeding application of beneficial genes that had previously been limited by the presence of undesirable linkage genes.
ACKNOWLEDGEMENTsThis study was supported by the Central Public Interest Scientific Institution Basal Research Fund of China National Rice Research Institute (Grant No. 2017RG002-4), the Agricultural Science and Technology Innovation Program of Chinese Academy of Agricultural Sciences, and Science, Technology and Innovation Committee of Shenzhen Municipality (Grant Nos. JCYJ20170303154506881 and JCYJ20170412155447658).
SUPPLEMENTAL DataThe following materials are available in the online version of this article at http://www.sciencedirect.com/science/journal/16726308; http://www.ricescience.org.
Supplemental File 1. Materials and methods used in this study.
Supplemental Table 1. Mutations detected in putative CRISPR/ Cas9 off-target sites.
Supplemental Table 2. Primers used in this study.
Supplemental Fig. 1. Isolation of possible transgene-free T1 plants from osc1-2.
Supplemental Fig. 2. Characterization of agronomic traits of wild type (WT) 4008S and osc1-2 plants.
Supplemental Fig. 3. Pairwise linkage disequilibrium heatmap (LD heatmap) in the Chalk5 and GS5 genes.
MATERIALS AND METHODSPlant materials and growth conditionsThe transgenic plants were generated from the japonica wide-compatibility photosensitive genic male-sterile line 4008S. All of the plants were grown under outdoor conditions in experimental fields in the China National Rice Research Institute, Fuyang, Hangzhou, China. Field management was conducted in accordance with normal agricultural practices.
Design of target sitesWe designed the target site of the chromogen C gene OsC1 according to a previous study (Shen et al, 2016). The sequence CCCATGGCGAAGGCAAGTGGCGC was selected as target site for editing of OsC1. We obtained the PCR products, including the target segment of OsC1in 4008S by using the primers OsC1-JC-F/R (Supplemental Table 1).
Plasmid construction and rice transformationThe pC1300-Cas9 binary vector carrying OsC1was constructed as previously described (Shen et al, 2019) and transformed into 4008S embryogenic calli via Agrobacterium-mediated transformation (strain EHA105) according to the method described by Hiei et al (1994). Transgenic lines were selected by culture in the presence of hygromycin (50 μ g/mL). All of the primers are listed in Supplemental Table 2.
Identification of transgenic positive strains and transgene-free plantsGenomic DNA of transgenic rice plants at the tillering stage was extracted using the cetyltrimethylammonium bromide (CTAB) method. Two specific primer pairs, hptII-F/hptII-R and Cas9-F/Cas9-R, in the expression vector were used to identify transgene-positive plants in the T0 generation (Supplemental Table 2). The two primer pairs were used to identify transgene-free plants in T1 and later generations, . Furthermore, a fragment of OsActin1, amplified using the primer pairs Actin-F/Actin-R, was used as a control to ensure that the quality of the genomic DNA was sufficient for PCR analysis.
Detection of mutations in T0 generationGenomic DNA of wild-type and transgenic rice plants at the tillering stage were extracted using the CTAB method. PCR amplification was performed with KOD FX DNA polymerase (Toyobo, Osaka, Japan) using primer pairs (Supplemental Table 2) surrounding the target sites. The PCR products were digested by restrict enzyme of NcoI and sequenced using the Sanger method, respectively. The sequences of transgenic plants were compared with those of the wild-type to detect mutations in the T0 generation.
Expression analysesTotal RNA was isolated from root and leaf of wild-type and osc1 plants using the AxyPerp Multisource Total RNA miniprep kit (Axygen, USA). Total RNA (1 μ g) was reverse-transcribed using a ReverTra Ace qPCR RT master mix with a gDNA remover kit (FSQ-301, Toyobo, Osaka, Japan). qRT-PCR was performed with gene-specific primers (Supplemental Table 2) using SYBR Green real time-PCR master mix (QPK-201, Toyobo, Osaka, Japan) and the ABI 7900 Fast Real Time-PCR System. Ubiquitin was used as an internal standard.
Linkage disequilibrium analysesThe linkage disequilibrium was evaluated within 132 japonica accessions that were obtained from the 3k single nucleotide polymorphism (SNP) database (Wang et al, 2018). The linkage disequilibrium heatmap (LD heatmap) was conducted using the R package (Shin et al, 2006).
Agronomic trait characterization4008S and osc1-2 plants were grown in the field under typical agricultural conditions. All agronomic traits, including plant height, tiller number, number of grains per panicle, seed-setting rate, 1 000-grain weight, average yield per plant, grain length, grain width, grain thickness, and grain length-to-width ratio were measured at the maturity. Ten plants were measured for each line. Differences were tested for significance using the Student’ s t-test (* , P < 0.05; * * , P < 0.01).
SUPPLEMENTARY REFERENCESHiei Y, Ohta S, Komari T, Kumashiro T. 1994. Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J, 6(2): 271-282.
Shen L, Wang C, Fu Y P, Wang J J, Liu Q, Zhang X M, Yan C J, Qian Q, Wang K J. 2016. QTL editing confers opposing yield performance in different rice varieties. J Integr Plant Biol, 60(2):89-93.
Shen L, Dong G J, Zhang Y, Hu G C, Zhang Q, Hu G L, Xu B, Ren D Y, Hu J, Zhu L, Gao Z Y, Zhang G H, Guo L B, Zeng D L, Qian Q. 2019. Rapid creation of new photoperiod-/thermo- sensitive genic male-sterile rice materials by CRISPR/Cas9 System. Rice Sci, 26(2): 129-132.
Shin J H, Blay S, McNeney B, Graham J. 2006. LDheatmap: An R function for graphical display of pairwise linkage disequilibria between single nucleotide polymorphism. J Stat Soft, 16: Code Snippet 3.
Wang W S, Mauleon R, Hu Z Q, Chebotarov D, Tai S S, Wu Z C, Li M, Zheng T Q, Fuentes R R, Zhang F, Mansueto L, Copetti D, Sanciangco M, Palis K C, Xu J L, Sun C, Fu B Y, Zhang H L, Gao Y M, Zhao X Q, Shen F, Cui X, Yu H, Li Z C, Chen M L, Detras J, Zhou Y L, Zhang X Y, Zhao Y, Kudrna D, Wang C C, Li R, Jia B, Lu J Y, He X C, Dong Z T, Xu J B, Li Y H, Wang M, Shi J X, Li J, Zhang D B, Lee S, Hu W S, Poliakov A, Dubchak I, Ulat V J, Borja F N, Mendoza J R, Ali J, Li J, Gao Q, Niu Y C, Yue Z, Naredo M E B, Talag J, Wang X Q, Li J J, Fang X D, Yin Y, Glaszmann J, Zhang J W, Li J Y, Hamilton R S, Wing R A, Ruan J, Zhang G Y, Wei C C, Alexandrov N, McNally K L, Li Z K, Leung H. 2018. Genomic variation in 3, 010 diverse accessions of Asian cultivated rice. Nature, 557(7703): 43-49.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 , Qian Qian
, Qian Qian