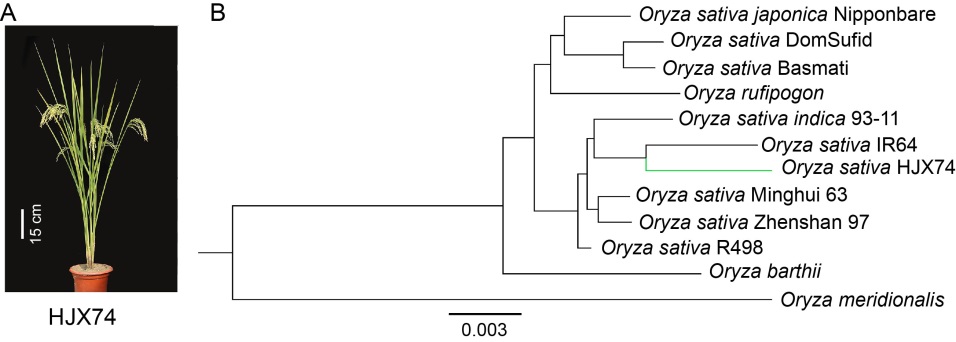
Rice (Oryza sativa L.) is grown nearly worldwide and provides the staple food for more than half of the global population (Luo et al, 2017). The genomes of several cultivated rice varieties including Nipponbare (NPB) (Kawahara et al, 2013; Sakai et al, 2013), IR64 (Tanaka et al, 2020), 93-11 (Zhang et al, 2018) and R498 (Du et al, 2017) at chromosome level, and Minghui 63 and Zhenshan 97 (Zhang et al, 2016) at scaffold level have been assembled, annotated and released, among which the R498 and NPB genomes are widely used as reference genomes in rice research. However, there are thousands of rice cultivars, landraces and wild rice varieties in the world with dramatically different genetic backgrounds, and the genomes of native rice varieties in South China, which is one of the major rice production areas in China, have not been de novo assembled. Huajingxian 74 (HJX74) is an indica rice variety bred in South China Agricultural University, Guangdong Province with widely environmental adaptability and high yield (www.ricedata.cn/ variety/varis/602548.htm). HJX74 exhibits significant phenotypic and genetic differences from those varieties whose whole genomes have been properly sequenced and assembled (Fig. 1).
In the past 30 years, a large library of single segment substitution lines (SSSLs) has been constructed using HJX74 as the receptor plant and 43 accessions that belong to 7 species of rice AA genome as donors. Hence, all these SSSLs are in the same genetic background (Zhang, 2019). The SSSL library has made a great contribution to the identification of QTLs/genes involved in disease resistance, fertility, panicle length, stress resistance, grain shape determination and so on (Wang S K et al, 2015; Fang et al, 2019; Wang et al, 2019). In addition, the SSSL library has provided a powerful platform for rice breeding by design (Luan et al, 2019; Zhao et al, 2019). The construction of a high-quality genome of the receptor parent (HJX74) of the SSSL library is therefore essential for improving the efficiency of rice genetic and mechanism studies for desirable agronomic traits, as well as accelerating the process of rice breeding by design. We produced a high-precision HJX74 chromosomal genome by performing whole-genome sequencing in the PacBio platform (Rhoads and Au, 2015), followed by the Hi-C-assisted assembly mount technology (van Berkum et al, 2010). The corresponding online platform has been constructed as well (https://RiceGenomicHJX.xiaomy.net). The sequence and de novo assembly of the HJX74 genome will significantly enrich the understanding of rice genome and provide a powerful tool for rice studies.
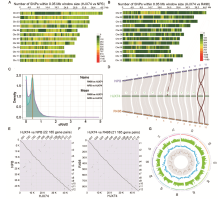
A total of 7 380 677 reads (137.31 Gb) of the HJX74 genome sequences were produced by PacBio SeqⅡ (Fig. 2-A and -B), and 51.23 Gb and 40.93 Gb of the sequence data were generated by common and Hi-C library preparation illumina sequencing, respectively. The overlapped group files (contig) consisting of 155 fasta format sequences with the size of 399.00 Mb (N50 = 14.41 Mb) (Table S1) were produced after being assembled and polished.
Table S1.
Table S1.
 Table S1. Comparison of contigs and scaffolds among Huajingxian 74 (HJX74) and two reference rice genomes (Nipponbare and R498).| Statistics without reference | HJX74 (contig level) | HJX74 | Nipponbare | R498 |
|---|
| Contigs | 155 | 108 | 63 | 14 | | Contigs ≥ 1 000 bp | 155 | 108 | 63 | 14 | | Contigs ≥ 5 000 bp | 141 | 94 | 62 | 14 | | Contigs ≥ 10 000 bp | 135 | 88 | 51 | 14 | | Contigs ≥ 25 000 bp | 126 | 80 | 20 | 14 | | Contigs ≥ 50 000 bp | 109 | 69 | 18 | 14 | | Largest contig | 26 609 203 | 44 230 994 | 43 270 923 | 44 361 539 | | Total length | 399 000 942 | 398 866 991 | 375 049 285 | 390 983 850 | | Total length ≥ 1 000 bp | 399 000 942 | 398 866 991 | 375 049 285 | 390 983 850 | | Total length ≥ 5 000 bp | 398 955 832 | 398 821 873 | 375 045 049 | 390 983 850 | | Total length ≥ 10 000 bp | 398 920 023 | 398 786 031 | 374 960 283 | 390 983 850 | | Total length ≥ 25 000 bp | 398 753 016 | 398 632 314 | 374 514 817 | 390 983 850 | | Total length ≥ 50 000 bp | 398 046 761 | 398 182 493 | 374 450 782 | 390 983 850 | | N50 | 14 408 600 | 31 354 834 | 29 958 434 | 31 778 392 | | N75 | 11 330 867 | 30 528 296 | 28 443 022 | 29 952 003 | | L50 | 11 | 6 | 6 | 6 | | L75 | 18 | 9 | 9 | 9 | | GC (%) | 43.7 | 43.7 | 43.57 | 43.62 | | # N’ s (Mismatches) | 0 | 5 511 | 118 258 | 50 000 | | # N’ s per 100 kbp (Mismatches) | 0 | 1.38 | 31.53 | 12.79 |
| Table S1. Comparison of contigs and scaffolds among Huajingxian 74 (HJX74) and two reference rice genomes (Nipponbare and R498). |
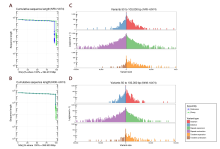
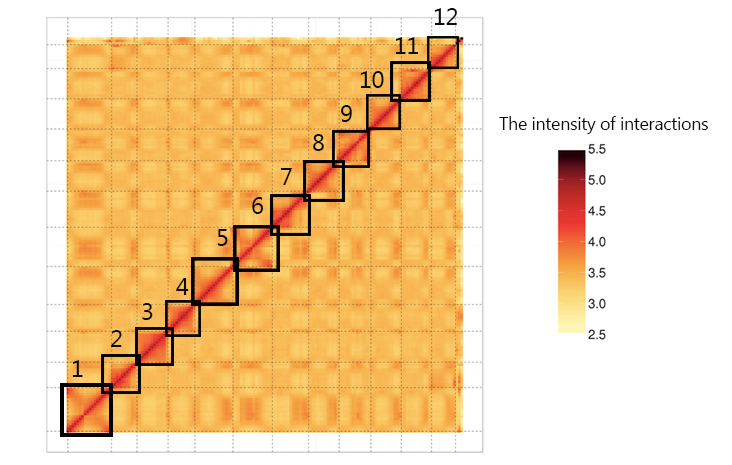
Visualization of the Hi-C signals indicated that 12 square matrix areas in the Hi-C heat map displayed significant differences from the background signal corresponding to the chromosome number of the rice nuclear genome (Fig. S1). The final polished scaffold genome was constructed by the Hi-C data and the consensus sequence file spanned 398.87 Mb, and there were 108 contigs for HJX74 including 12 chromosome lengths contigs (Fig. 2-D and Table S1). The genome assemblies recovered more than 98% of the 1 440 Benchmarking Universal single- copy orthologs (BUSCO) embryophyte genes and completely assembled more than 92.5% of the 248 embryophyte core genes from the Core Eukaryotic Genes Mapping Approach (CEGMA) database (Li et al, 2020) (Table S2). Long terminal repeat-retotransposons (LTR-RTs) assembly index (LAI) of the HJX74 genome was calculated to be 23.42, which is close to the high-quality rice genome of NPB (22.59) and R498 (23.94) (Table S3).
Table S2.
Table S2.
Table S2. Evaluation of Huangjingxian 74 genome assembly by Benchmarking Universal single-copy ortholog (BUSCO) and Core Eukaryotic Genes Mapping Approach (CEGMA).| Method (Database) | Data |
|---|
| BUSCO | C, 98.3%; S, 97.6%, D, 0.7%; F, 0.3%; M, 1.4%; n, 1440 | | CEGMA | Complet: 230 (92.74%); Complete + Partial: 235 (94.76%) |
C, Complete single-copy BUSCOs; D, Complete duplicated BUSCOs; F, Fragmented BUSCOs; M, Missing BUSCOs; n, Total BUSCO groups searcheds; Complete, Core gene > 70% assembled; Complete + partial, The partial assembly of core gene. | Table S2. Evaluation of Huangjingxian 74 genome assembly by Benchmarking Universal single-copy ortholog (BUSCO) and Core Eukaryotic Genes Mapping Approach (CEGMA). |
Table S3.
Table S3.
Table S3. LTR-TRs assembly index (LAI) of R498, Nipponbare and Huajingxian 74 (HJX74).| Species | Length | Intact | Total | Raw LAI | LAI |
|---|
| R498 (indica) | 390 983 850 | 0.0663 | 0.2828 | 23.44 | 23.94 | | Nipponbare (japonica) | 375 049 285 | 0.0505 | 0.2452 | 20.58 | 22.59 | | HJX74 (indica) | 398 866 991 | 0.0656 | 0.2887 | 22.73 | 23.42 |
| Table S3. LTR-TRs assembly index (LAI) of R498, Nipponbare and Huajingxian 74 (HJX74). |
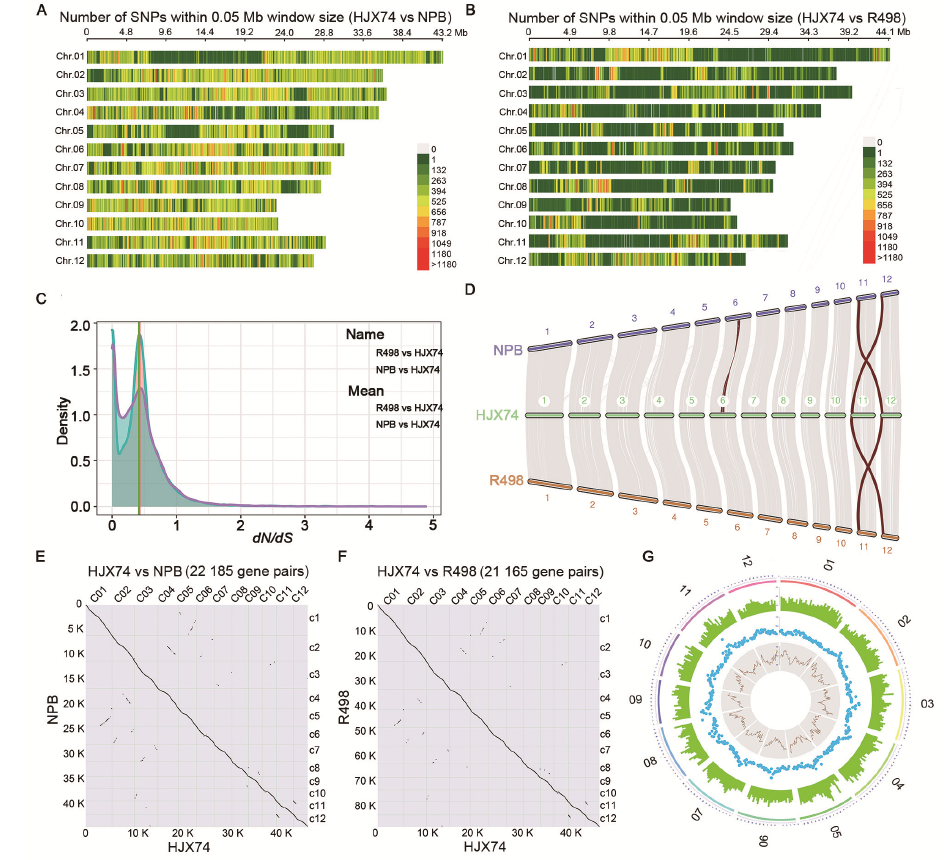
Combining ab initio, protein and expressed sequence tag (EST) evidences with consensus gene prediction (Zhang et al, 2015), we annotated the HJX74 genome with 46 993 non-redundant genes. Among them, 39 002 genes (83.0%) form 27 202 clusters with genes from 11 otherOryza species, whereas 7 991 genes present singletons in the OrthoVenn2 (Wang Y et al, 2015). The clustering analysis based on Markov Clustering (MCL) algorithm indicates high annotation reliability. Totally 2 850 single-copy gene clusters were generated by the orthologous cluster analysis of direct homology in 9 Oryza sativa varieties, O. rufipogon, O. meridionalis and O. barthii on the platform OthoVenn2 (Table S4). The phylogenetic tree constructed by using the coding region nucleic acid sequence of 2 850 single- copy lineal homologous gene clusters indicated that HJX74 was clustered in the clade of O. sativasubsp. indica and had the closest genetic relationship with IR64 (Fig. 1-B and Table S5). HJX74 was genetically far from NPB and R498, even HJX74 and R498 were clustered within the indica rice clade, which is consistent with the SNPs, InDels and persence and absence variations (PAVs) across the 12 chromosomes in HJX74 compared to NPB and R498 (Fig. 2-A and -B; Fig. S2 and Tables S6 and S7). In addition, more genes were presented in HJX74/ R498 at a peak of 0.4-0.5 than HJX74/NPB from the density curve of dN/dS(Kryazhimskiy and Plotkin, 2008), which suggested more genes in HJX74 were positively selected when compared with R498 than the comparation with NPB (Fig. 2-C). The reason for this phenomenon is possibly due to the crossbreeding between rice subspecies (indica andjaponica) during the HJX74 breeding process and preference to tropical japonica as germplasm resources for rice breeding in South China.
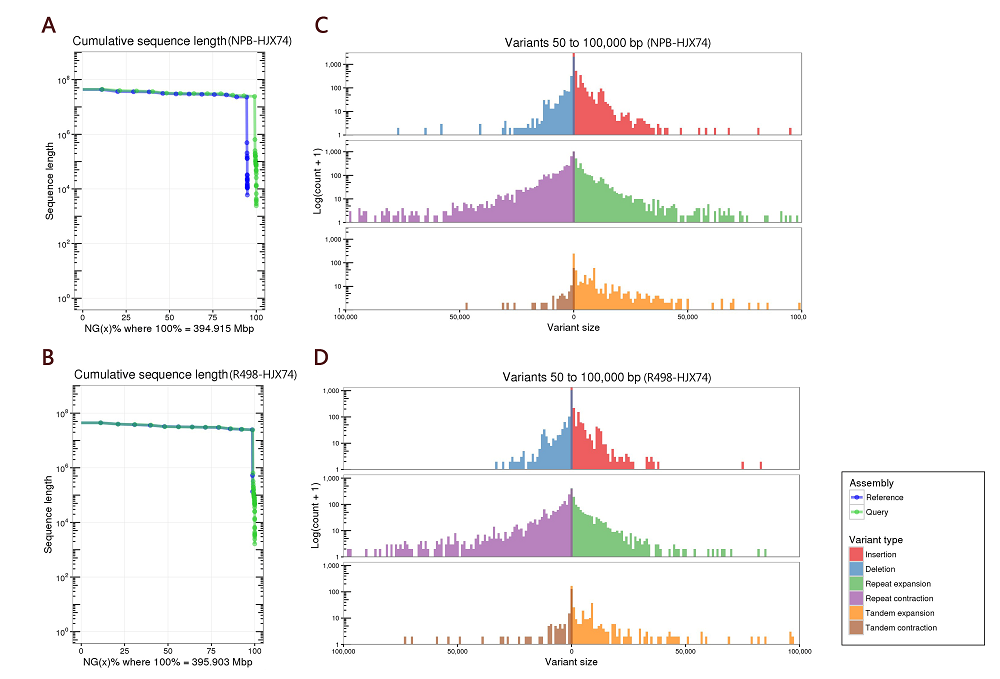
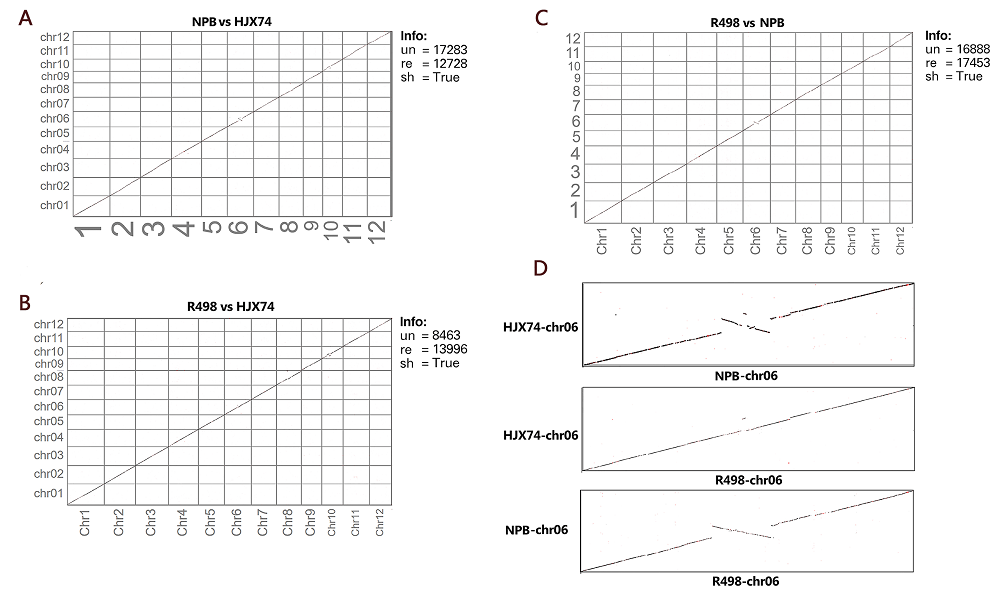
The relative lengths of HJX74 chromosomes are consistent with NPB and R498 (Table S3). According to the whole- genome comparison, the genome of HJX74, at the position of about 12-17 Mb on chromosome 6, showed a sequence inversion with a length of about 5 Mb compared with the NPB genome, while the HJX74 sequence was in the same order as R498 (Fig. S3). Besides, the HJX74 genome was nearly 8.1 Mb and 25.2 Mb larger than R498 (390.9 Mb) and NPB (373.8 Mb), respectively. We performed a whole-genome comparison to examine the synteny between the HJX74 and R498/NPB genomes using the python version program MCScanX (Wang et al, 2012). HJX74 showed a high degree of synteny and the same large inversion in the middle of chromosome 6 with indica/japonica genomes, which was consistent with the whole- genome alignment between the HJX74 and R498/NPB genomes (Fig. 2-D to -F and Fig. S3). This phenomenon or the disordered alignment to NPB in the same locus was also respectively detected in the genomes of O. sativa, Basmati 334 and DomSufid (Choi et al, 2020; Xie et al, 2020). This long fragment staining inversion phenomenon existed in this site indeed, which suggested that the inversion might have been occurred during the process of rice subspecies differentiation. There is a about 3 Mb large-scale syntenic block between the short arms of chromosomes 11 and 12 according to the synteny plot, which was estimated to result from a duplication event 7.7 million years ago and was consistent with previous research (The Rice Chromosomes 11 and 12 Sequencing Consortia, 2005).
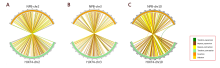
There are a considerable number of PAVs between the genomes of HJX74 and NPB (Table S8 and Fig. S2-B). Compared with NPB, the HJX74 genome has more long-fragment insertion sequences and repeated fragment expansions (Fig. S2-B). Three NPB chromosomes (NPB-Chr.02, NPB-Chr.03 and NPB-Chr.10) with the greatest difference from HJX74 were compared. The long-term insertions (> 10 kb) and tandem/repeats contributed significantly to the longer chromosome length of HJX74 compared to NPB (Fig. S4-A to -C). This result tallies with the previous report that the chromosome length difference was most probably due to the changes in tandem/repeat regions (Kim et al, 2017). In contrast, the length of each chromosome of HJX74 was close to that of R498 with an average length difference about 0.075 Mb (Table S9).
Table S7.
Table S7.
Table S7. Clustering of homologous genes of 12 species of rice.| Species | Protein | Cluster | Singleton |
|---|
| Oryza barthii | 34 575 | 24 966 | 3 223 | | Oryza meridionalis | 29 308 | 20 721 | 4 088 | | Oryza rufipogon | 37 062 | 27 114 | 2 912 | | Oryza sativaNipponbare | 35 667 | 23 642 | 5 446 | | Oryza sativa93-11 | 37 878 | 25 644 | 3 690 | | Oryza sativaBasmati | 41 259 | 29 417 | 4 210 | | Oryza sativaDomSufid | 38 329 | 28 037 | 3 490 | | Oryza sativaMinghui 63 | 83 258 | 40 508 | 13 514 | | Oryza sativaR498 | 88 169 | 27 111 | 33 193 | | Oryza sativaZhenshan 97 | 84 957 | 40 646 | 14 163 | | Oryza sativaHuajingxian 74 | 46 933 | 27 202 | 7 991 | | Oryza sativaIR64 | 41 458 | 23 591 | 9 810 |
| Table S7. Clustering of homologous genes of 12 species of rice. |
Table S9.
Table S9.
Table S9. Chromosome lengths of Huajingxian 74 (HJX74), Nipponbare (NPB) and R498.| Chromosome | Length of each chromosome (bp) |
|---|
| HJX74 | NPB | R498 |
|---|
| 1 | 44 230 993 | 43 270 923 | 44 361 539 | | 2 | 38 415 586 | 35 937 250 | 37 764 328 | | 3 | 39 700 684 | 36 413 819 | 39 691 490 | | 4 | 36 567 208 | 35 502 694 | 35 849 732 | | 5 | 30 862 635 | 29 958 434 | 31 237 231 | | 6 | 31 992 588 | 31 248 787 | 32 465 040 | | 7 | 30 655 926 | 29 697 621 | 30 277 827 | | 8 | 30 528 296 | 28 443 022 | 29 952 003 | | 9 | 24 203 274 | 23 012 720 | 24 760 661 | | 10 | 25 662 364 | 23 207 287 | 25 582 588 | | 11 | 31 354 834 | 29 021 106 | 31 778 392 | | 12 | 27 052 960 | 27 531 856 | 26 601 357 | | Average difference | | 1.390 Mb | 0.07543 Mb |
| Table S9. Chromosome lengths of Huajingxian 74 (HJX74), Nipponbare (NPB) and R498. |
Then, we found that the LTR-RT length and type ratio (Gypsy/ Copia/unknown) of the HJX74 genome were similar to those of R498, but significantly different from those of NPB (Table S10). Previous research reported that the two subspecies of rice, indica and japonica, have experienced independent amplification or loss of LTR-RTs after the divergence (Du et al, 2017). In this study, the chromosome structure comparison showed fewer differences in PAVs and LTR-RTs between two indica varieties HJX74 and R498, but their PAVs and LTR-RTs were very different from those of NPB. Meanwhile, a total of 26 647 simple sequence repeat loci, with the number of repeating units ≥ 3 bp, were detected in 12 chromosomes of HJX74 (Fig. 2-G and Table S11), which demonstrated the promising application of the HJX74 genome in the development of molecular breeding markers.
To encourage the use of the genome of HJX74 and other rice varieties, a platform (https://RiceGenomicHJX.xiaomy.net) supporting sequence search (Blast), gene browse, download and extraction were built with the support from the Guangdong Provincial Key Laboratory of Plant Molecular Breeding, China. The platform also collects information about the mutation sites in HJX74 and other rice genomes, and multiple rice research platforms and websites. Further improvement and development of the platform is underway to optimize its application (Fig. S5).
Table S10. Repeat content (LTR-RTs) in Huajingxian 74 (HJX74), R498 and Nipponbare (NPB).| Repeat content | HJX74 | R498 | NPB |
|---|
| Length (bp) | Percentage of genome (%) | Length (bp) | Percentage of genome (%) | Length (bp) | Percentage of genome (%) |
|---|
| Total | 110 814 118 | 27.78 | 109 473 086 | 28.00 | 90 414 977 | 24.12 | | Gypsy | 84 080 899 | 21.08 | 82 619 222 | 21.13 | 64 570 822 | 17.22 | | Copia | 13 457 780 | 3.37 | 12 661 044 | 3.24 | 13 948 467 | 3.72 | | Unknown | 13 275 439 | 3.33 | 14 192 820 | 3.63 | 11 895 688 | 3.17 |
| Table S10. Repeat content (LTR-RTs) in Huajingxian 74 (HJX74), R498 and Nipponbare (NPB). |
In previous studies, considerable progress has been made by combining bioinformatics and whole genome sequencing methods (such as RNA-seq and genome-wide association study) with traditional molecular biology methods for germplasm resource mining and molecular breeding in rice (Shao et al, 2019; Groen et al, 2020). However, these technologies require a reliable reference genome. Here, we presented a highly contiguous and near-complete genome assembly for HJX74, a high-yielding indica rice variety widely-grown in South China. As a platform variety, HJX74 has been implemented to construct a large SSSL library with 2 360 independent lines (Zhang, 2019). The SSSL library has an excellent application prospect in rice breeding by design and QTL/gene identifications (Zhou et al, 2017). Compared with NPB, the utilization of the HJX74 reference genome is able to detect more SNP loci or insertion/deletion sites in many PAVs while combining with whole genome sequencing technologies (Fig. S6). Our work provides a precise reference genome and an accessible utilization platform for further research based on the SSSL library. There is no doubt that this reference genome of the receptor parent of the SSSL library will contribute to simplifying the mining and identification processes of rice functional genes controlling agronomic traits of interest, thereby promoting the research and application of rice breeding by design.
AcknowledgEmentsThis study was supported by the National Key Research and Development Program of China (Grant No. 2016YFD0100406), National College Students Innovation and Entrepreneurship Foundation of China (Grant No. 201910564054), National Natural Science Foundation of China (Grant Nos. 91735304 and 31622041) and Special Project for Leading Talents in Innovation of Science and Technology of Guangdong Province, China (Grant No. 2016TX03N224). We thank Ji Zhe (Department of Plant Sciences, University of Oxford) for suggestions.
MethodsCollection of rice samples and DNA extractions
Plant materials were planted at the experimental station at the South China Agricultural University in Guangdong Province, China (23º 10′ 3.07′ ′ N, 113º 21′ 41.39′ ′ E). The seeds used for germination were produced from flowers that were bagged to prevent cross-pollination, and harvested in the late season of 2012. After hydroponic at 28 º C, plant tissue of about 2.0 g in weight was collected from two-week-old seedlings of Huangjingxian 74 (HJX74) for DNA extraction.
Genomic DNA was extracted by the CTAB method (Porebski et al, 1997). The quality of the extracted DNA library was determined by running on 1% agarose gel electrophoresis and using the QubitTM Fluorometer fluorescence platform. Specifically, only a single band of DNA should be present on the agarose gel, and the length of the band should be more than 30 kb. The concentration of the genomic DNA was 302 ng/µ L.
Library preparation and sequencing
SMRTbell Express Template Prep Kit 2.0 was used for library preparation of the HJX74 template and the platform Pacbio SeqⅡ was used for sequencing. Platform Illumina Hiseq was used to generate short fragment pair-end sequence (2 × 150). In the Hi-C experiment, the genomic DNA of HJX74 was fixed with 1% formaldehyde solution, before being digested by the DpnⅡ restriction enzyme. The binding to biotin markers was followed by using T4 DNA ligase (NEB, Ipswich, USA). DNA fragments labeled by biotin were finally separated on Dynabeads® M-280 Streptavidin (Thermo Fisher Scientific , Waltham, USA). Hi-C libraries were sequenced and quality controlled on an Illumina Hiseq X Ten sequencer (Illumina, San Diego, USA).
Genome assembly, polishing and scaffolding
PacBio reads were assembled by Canu 2.0 (corOutCoverage = 120, corMinCoverage = 2, minReadLength = 2000, minOverlapLength = 500). To improve the accuracy of the result, the preliminary.config file were subsequently polished using Pacbio reads by Quiver (https://github.com/PacificBiosciences/GenomicConsensus) and illumina reads by Pilon v2.0 (https://github.com/broadinstitute/pilon) and NextPolish (Hu et al, 2019).
Short Hi-C reads were mapped by BWA mem (Li, 2013), Samtools v1.9 (Li et al, 2009) and GATK software (https://github.com/broadinstitute/gatk/). HiC-Pro (Servant et al, 2015) and HiCplotter (Akdemir and Chin, 2015) were used for the visualization of the Hi-C interaction signals. HapCUT (Bansal and Bafna, 2008) algorithm and HICUP pipeline (Wingett et al, 2015) were respectively used for haplotype reconstructing and generating contact matric based on the Hi-C data. The integrity of the assembled genome was assessed by Benchmarking Universal single-copy orthologs (BUSCO) and Core Eukaryotic Genes Mapping Approach (CEGMA) (Li et al, 2020It is not listed in REFERENCE. Please supply.).
Gene annotation
A combined strategy was adopted to predict the protein-coding genes in HJX74 genome. Augustus v3.0.3 (Stanke et al, 2004) was used to detect the hypothetical gene-coding regions in the HJX74 genome (ab initio). We developed a Pipline called RiceOrthoblast (https://github.com/lipingfangs/ RiceOrthoblast) by intergrating Software BLAST+ v2.9.0 (ftp://ftp.ncbi.nlm.nih.gov/blast/ executables/blast+/) and Genewise v2.4.1 (http://www. ebi.ac.uk/~birney/wise2/) to annotate the HJX74 genome with genomic homology from the reference protein sequence file (Oryza_sativa. IRGSP-1.0.pep.all.fa; ftp://ftp.ensemblgenomes.org/ pub/plants/release-47/fasta/ oryza_sativa/pep/), which generate the non-redundant homologous gene annotation file (GFF3; Pep) for lineal homology phylogenetic analysis as well as the protein evidence of further annotation. To further aid gene annotation, 21 682 489 Illumina RNA-seq reads were mapped to the HJX74 genome by Hisat2 v2.1.0 (Pertea et al, 2016) and samtools v1.9. The .bam file generated above was assembled into transcripts and annotations files by Stringties v1.3.5 (Pertea et al, 2016) and TransDecoder v5.5.0 (Kim et al, 2016). EVidenceModeler (EVM) (Haas et al, 2008) was used to combine the predicted results from ab initio, protein and Expressed Sequence Tag (ESTPlease supply the full name.) evidences into consensus gene predictions.
Phylogenomic analysis
OrthoVenn2 (Wang et al, 2015It is not listed in REFERENCE. Please supply.) and its corresponding database were used for the comparison and clustering of nine cultivated rice samples [Nipponbare (NPB), 93-11, R498, IR64, Basmati, DomSuid, Zhenshan 97 (ZS97), Minghui 63 (MH63) and HJX74] and three wild rice samples (Oryza meridionalis, O. barthii and O. rufipogon). Protein sequences of single-copy gene clusters produced by direct homologous clustering of the 12 samples were aligned by MAFFT v7.0 (Katoh et al, 2002). Iqtree v1.1.6.1 (Nguyen et al, 2015) was used to match the optimal amino acid replacement model (Modelfinder module) and to build the phylogenetic tree.
Repetitive DNA annotation
The LTR_finder software (Xu and Wang, 2007) was used for long terminal repeat (LTR) locus detection of HJX74 and three published cultivated rice genomes (NPB, 93-11, R498). The result files were conveyed to LTR_retriever (Ou and Jiang, 2018) for LTR retrotransposons (LTR-RTs) site recognition, combined screening of the HJX74 genome and the calculation of LTR-RTs Assembly index (LAI).
MISA Please supply the full name.It is the full name of the software itself.(Thiel et al, 2003) was used for the detection of simple sequence repeats (SSR) of HJX74 (the number of repeating unit ≥ 3 bp was calculated). The GC content and the density of SSR and LTR-RTs were visualized by R package OmicCircos (Hu et al, 2014).
Genome-wide comparison
Mummer v4.0.0 (-maxmatch -l 100 -c 500) (http://mummer.sourceforge.net/) was used for the comparison of the HJX74 and three published cultivated rice genomes (NPB, R498Only two?Yes, only two cultivated rice were used in this analysis.). We examined and classified the presence/absence variants (PAVs) between these genomes by the computational packages assemblytics (Nattestad and Schatz, 2016It is not listed in REFERENCE. Please supply.). Using the python version of the software MCScanX (https://github.com/tanghaibao/jcvi/ wiki/MCscan-(Python-version)) with default parameters, we identified collinear orthologous genes, plotted the synteny blocks, and combined codeml programme of PAML Please supply the full name.It is the full name of the software itself.(Yang, 1997) to calculated the dN/dSvalues (Kryazhimskiy and Plotkin, 2008) for HJX74 and NPB, R498. The distribution of dN/dSvalues was plotted with R package ggplot2.
Platform construction
The platform (https://RiceGenomicHJX.xiaomy.net) was rooted in the Guangdong Provincial Key Laboratory of Plant Molecular Breeding, State Key Laboratory for Conservation and Utilization of Subtropical Agro- Bioresources, South China Agricultural. Its front-end kernel is based on HTML5, CSS, and the framework vue.js of Javascript. The back-end sequence search (blast) function implementation is based on the open-source PHP project Viroblast (https://indra.mullins.microbiol.washington.edu/viroblast/viroblast.php). The ssequence extraction software was realized by web pages as well as the attached tool FPtools developed by custom Python script.
Data availability
All analyses and quality control steps were coded in Python3 scripts or Linux shell commands/scripts except where stated explicitly. The custom Python3 codes for building overlap and generating final sequences are provided as https://github.com/lipingfangs. The sequence reads are available at The Genome Sequence Archive (GSA) (http://gsa.big.ac.cn/index.jsp) under project code PRJCA002801. The genome assembly of HJX74, and the data from whole genome sequence (WGS) sequencing have been deposited under NCBI BioProjects: PRJNA636594 (common illumina sequence data), PRJNA637189 (Pacbio data) and PRJNA637223 (Hi-C illumina sequence data). The accessions to the HJX74 assembly and the Illumina RNA-seq reads are PRJNA637414 and PRJNA639680, respectively. The assembled genome, annotated genes and protein files for HJX74, NPB, R498, 93-11, IR64, Basmati, DomSuid, MH63 and ZS97, and the data for displaying the example of sequence difference in PAVs locus are also accessible at https://RiceGenomicHJX.xiaomy.net.
REFERENCES
Akdemir K C, Chin L. 2015. HiCPlotter integrates genomic data with interaction matrices. Genome Biol, 16: 198.
Bansal V, Bafna V. 2008. HapCUT: An efficient and accurate algorithm for the haplotype assembly problem. Bioinformatics, 24: 153-159.
Haas B J, Salzberg S L, Zhu W, Haas M, Allen J E, Orvis J, White O, Buell R C, Wortman J R. 2008. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol, 9: R7.
Hu Y, Yan C H, Hsu C H, Chen Q R, Niu K, Komatsoulis G A, Meerzaman D. 2014. OmicCircos: A simple-to-use R package for the circular visualization of multidimensional omics data. Cancer Inform, 13: 13-20.
Hu, J., J. Fan, Z. Sun, S. Liu. 2019. NextPolish: a fast and efficient genome polishing tool for long read assembly. Bioinformatics 36: 2253-2255.
Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: A novel method for NPB id multiple sequence alignment based on fast Fourier transform. Nucl Acids Res, 30: 3059-3066.
Kim H, Hwang D, Lee B, Park J C, Lee Y H, Lee J. 2016. De novo assembly and annotation of the marine mysid (Neomysis awatschensis) transcriptome. Mar Genom, 28: 41-43.
Kryazhimskiy S, Plotkin J B. 2008. The population genetics of dN/dS.PLoS Genet, 4: e1000304.
Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Genomics, 1303: 1-3.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinformatics, 25: 2078-2079.
Li, W., K. Li, Q. Zhang, T. Zhu, Y. Zhang, C. Shi, Y. Liu, E. Xia, J. Jiang, C. Shi, L. Zhang, H. Huang, Y. Tong, Y. Liu, D. Zhang, Y. Zhao, W. Jiang, Y. Zhao, S. Mao, L. Gao. 2020. Improved hybrid de novo genome assembly and annotation of African wild rice, Oryza longistaminata, from Illumina and PacBio sequencing reads. The Plant Genome 13:e20001.
Nguyen L, Schmidt H A, von Haeseler A, Minh B Q. 2015. IQ-TREE: A fast and effective stochastic Algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol, 32: 268-274.
Nattestad M, Schatz M C. 2016. Assemblytics: a web analytics tool for the detection of variants from an assembly. Bioinformatics 32: 3021-3023.
Ou S, Jiang N. 2018. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol, 176: 1410-1422.
Pertea M, Kim D, Pertea G M, Leek J T, Salzberg S L. 2016. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat Protoc, 11: 1650-1667.
Porebski S, Bailey L G, Baum B R. 1997. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol Biol Rep, 15: 8-15.
Servant N, Varoquaux N, Lajoie B R, Viara E, Chen C J, Vert J P, Heard E, Dekker J, Barillot E. 2015. HiC-Pro: An optimized and flexible pipeline for Hi-C data processing. Genome Biol, 16: 259.
Stanke M, Steinkamp R, Waack S, Morgenstern B. 2004. AUGUSTUS: A web server for gene finding in eukaryotes. Nucl Acids Res, 32: W309-W312.
Thiel T, Michalek W, Varshney R, Graner A. 2003. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet, 106: 411-422.
Wang, Y., D. Coleman-Derr, G. Chen, Y. Gu. 2015. OrthoVenn: a web server for genome wide comparison and annotation of orthologous clusters across multiple species.Nucl Acids Res, 43: W78-84.
Wingett S W, Ewels P, Furlan-Magaril M, Nagano T, Schoenfelder S, Fraser P, Andrews S. 2015. HiCUP: Pipeline for mapping and processing Hi-C data. F1000 Res, 4: 1310.
Xu Z, Wang H. 2007. LTR_FINDER: An efficient tool for the prediction of full-length LTR retrotransposons. Nucl Acids Res, 35: W265-W268.
Yang Z. 1997. PAML: A program package for phylogenetic analysis by maximum likelihood. Computer Appl Biosci, 13: 555-556.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 ]
]